


Search
© 2024 khagapharmacy.com
NEUROSCIENCE
Contents
Creutzfeldt Jakob Disease: The Silent KillerIntroduction to Creutzfeldt Jakob Disease (CJD):What is Creutzfeldt Jakob Disease (CJD)?:Table of ContentsBrief History of Creutzfeldt Jakob Disease:Types of Creutzfeldt Jakob Disease:Causes of Creutzfeldt Jakob Disease:Symptoms of Creutzfeldt Jakob Disease:Diagnosis of Creutzfeldt Jakob Disease:Treatment of Creutzfeldt Jakob Disease:What is Prion Diseases:How Prions Cause Disease:FAQs about Creutzfeldt Jakob Disease: Conclusion:Leave a Reply Cancel reply
Creutzfeldt Jakob Disease: The Silent Killer
-
 Rahul Priydarss
Rahul Priydarss
Creutzfeldt Jakob Disease (CJD) is a rare, fatal neurodegenerative disorder caused by abnormal prion proteins. Affecting adults primarily, CJD leads to rapid mental and physical deterioration, characterized by severe neurological symptoms and death within a year of onset. The disease exists in several forms: sporadic, hereditary, and acquired. Sporadic CJD occurs without known cause, hereditary CJD results from genetic mutations, and acquired CJD arises from exposure to infectious prions. Diagnosis involves clinical assessment, cerebrospinal fluid analysis, EEG, MRI, and, occasionally, genetic testing. While there is no cure, ongoing research aims to improve understanding, diagnostics, and treatment.
- Advertisement -
Introduction to Creutzfeldt Jakob Disease (CJD):
Creutzfeldt Jakob Disease (CJD) is a rare, degenerative, and fatal brain disorder that primarily affects adults. It is one of several transmissible spongiform encephalopathies (TSEs) and is characterized by rapid mental deterioration, leading to severe neurological symptoms and, ultimately, death. There are several forms of CJD, including sporadic, hereditary, and acquired variants. The disease is caused by abnormal proteins called prions, which cause brain tissue to become damaged and spongy. Although CJD is rare, it has significant implications for affected individuals and their families, making increased awareness and research into the disease critical.
What is Creutzfeldt Jakob Disease (CJD)?:
Creutzfeldt Jakob Disease (CJD) is a rare and fatal neurodegenerative disorder that rapidly deteriorates brain function. It belongs to a group of conditions known as transmissible spongiform encephalopathies (TSEs), which cause brain tissue to develop a sponge-like appearance. The disease is caused by abnormal, misfolded proteins called prions that lead to brain damage. CJD can manifest in several forms, including sporadic, hereditary, and acquired types, each with distinct origins. Although the disease is uncommon, it progresses quickly, leading to severe neurological symptoms and death, usually within a year of onset.

Table of Contents
Brief History of Creutzfeldt Jakob Disease:
Creutzfeldt Jakob Disease (CJD) was first described in the early 20th century by two German neurologists, Hans Gerhard Creutzfeldt and Alfons Jakob. In 1920, Creutzfeldt reported the case of a young woman with progressive mental and motor deterioration, while Jakob described several cases with similar symptoms and pathological findings a year later. Initially, the disease was thought to be a rare form of encephalitis.
Over the decades, further research revealed that CJD was caused by abnormal prion proteins, leading to the classification of the disease as a transmissible spongiform encephalopathy (TSE). The identification of different forms of CJD, including sporadic, hereditary, and acquired variants, expanded the understanding of the disease’s transmission and pathogenesis. Notably, the variant CJD (vCJD) form was identified in the 1990s, linked to the consumption of beef from cattle affected by bovine spongiform encephalopathy (BSE), also known as mad cow disease. This discovery prompted significant public health measures to control the spread of prion diseases.
- Advertisement -
Types of Creutzfeldt Jakob Disease:
Creutzfeldt Jakob Disease (CJD) can be categorized into several distinct types based on its causes and characteristics. The three primary forms of CJD are sporadic, hereditary, and acquired.
Sporadic CJD (sCJD) is the most common form, accounting for about 85% of all CJD cases. The exact cause of sCJD remains unknown, and it appears to occur spontaneously without any known risk factors. This form of the disease typically affects individuals around the age of 60 and progresses rapidly, often leading to death within a year of symptom onset. The sudden and unexplained nature of sporadic CJD makes it particularly challenging to diagnose and understand.
Hereditary CJD (hCJD) accounts for about 10-15% of all CJD cases. This form of the disease is caused by mutations in the PRNP gene, which provides instructions for making prion protein. Hereditary CJD is inherited in an autosomal dominant manner, meaning that a single copy of the mutated gene from either parent can cause the disease. Symptoms of hereditary CJD often appear earlier than in sporadic CJD, and the progression of the disease can vary widely among affected individuals.
Acquired CJD is a rare form of the disease that results from exposure to infectious prions through external sources. This category includes iatrogenic CJD (iCJD) and variant CJD (vCJD). Iatrogenic CJD is caused by medical procedures, such as contaminated surgical instruments, corneal transplants, or growth hormone injections derived from human pituitary glands. Variant CJD (vCJD) is linked to consuming beef from cattle with bovine spongiform encephalopathy (BSE), also known as mad cow disease. First identified in the 1990s, vCJD affects younger individuals and has a longer duration of illness compared to other forms of CJD.
- Advertisement -
Causes of Creutzfeldt Jakob Disease:
Creutzfeldt Jakob Disease (CJD) is a rare and fatal degenerative brain disorder. It is caused by an abnormal infectious protein called prion. Here are the key causes.
Spontaneous Mutation: In most cases, CJD occurs spontaneously without any known cause. This is called sporadic CJD and accounts for about 85% of cases.
Genetic Mutation: Some people inherit a mutated gene that predisposes them to CJD. This is known as familial or genetic CJD, which accounts for about 10-15% of cases.
Acquired Infection: CJD can also be acquired through exposure to contaminated tissues or medical procedures. This includes:
- Iatrogenic Transmission: Transmission via medical procedures involving contaminated surgical instruments, transplants (like corneal transplants), or injections of contaminated growth hormone.
- Variant CJD (vCJD): This form is linked to consumption of beef contaminated with Bovine Spongiform Encephalopathy (BSE), commonly known as mad cow disease. Variant CJD is rare but has been linked to a specific prion strain.
Unknown Factors: In a small number of cases, the cause of CJD remains unknown, even after a thorough investigation.
- Advertisement -
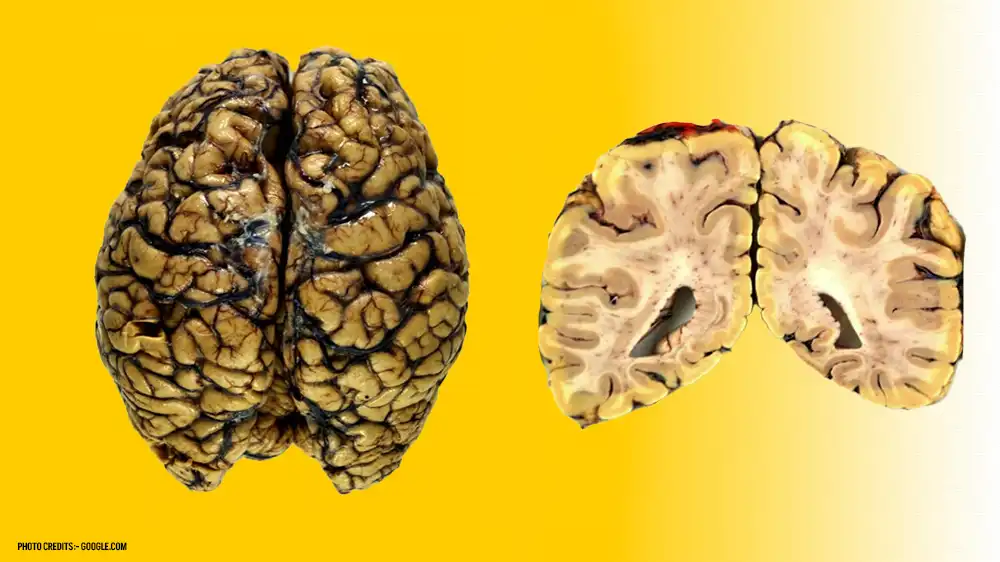
Symptoms of Creutzfeldt Jakob Disease:
Creutzfeldt Jakob Disease (CJD) manifests with a range of neurological symptoms that progressively worsen over time. Here are the key symptoms, each explained.
Cognitive Decline: Early symptoms often include memory problems, changes in thinking abilities, and difficulties with concentration and attention. This decline in cognitive function can lead to confusion and disorientation.
Behavioral Changes: Patients may experience rapid mood swings, personality changes, and uncharacteristic behaviors. These changes can range from apathy and withdrawal to agitation and aggression.
Muscle Coordination Problems: As the disease progresses, individuals may develop problems with coordination and balance. This can lead to stumbling, difficulty walking, and jerky movements (myoclonus).
Visual Disturbances: Some patients may experience visual disturbances such as blurred vision or hallucinations.
Sleep Disturbances: Insomnia or other sleep disturbances are common in the later stages of the disease.
Physical Symptoms: As CJD advances, muscle stiffness, tremors, and difficulty swallowing (dysphagia) may occur. Patients may also become increasingly immobile and require assistance with daily activities.
Dementia: Severe dementia develops as the disease progresses, leading to a profound loss of intellectual abilities and memory. Communication becomes increasingly challenging, and patients may lose the ability to recognize family members or perform basic tasks.
End-Stage Symptoms: In the final stages of CJD, patients typically enter a state of profound disability, with minimal responsiveness and complete dependence on caregivers. The disease is ultimately fatal, usually within months to a few years after symptoms first appear.
- Advertisement -
Diagnosis of Creutzfeldt Jakob Disease:
Diagnosing Creutzfeldt Jakob Disease (CJD) can be challenging due to its rarity and the similarity of its symptoms to other neurological conditions. A thorough diagnostic process typically involves the following steps.
Clinical Assessment: A detailed medical history and neurological examination are conducted to assess symptoms, their progression, and any potential risk factors (such as recent surgeries or family history).
Cerebrospinal Fluid (CSF) Analysis: A lumbar puncture (spinal tap) may be performed to analyze the CSF for elevated levels of 14-3-3 protein and tau protein. These proteins are often elevated in CJD but can also be present in other neurological disorders.
Electroencephalogram (EEG): EEG measures the electrical activity in the brain. In CJD, characteristic periodic sharp wave complexes (PSWCs) may be seen, though they are not specific to CJD and can also be found in other conditions.
MRI Brain Imaging: MRI scans can show specific changes in the brain associated with CJD, such as cortical ribboning (a pattern of increased signal intensity in the cerebral cortex) and brain atrophy. These findings can support a diagnosis but are not definitive on their own.
Biopsy or Autopsy: A brain biopsy (rarely done due to its invasive nature) or autopsy after death can provide conclusive evidence of CJD through the presence of prion protein accumulation and characteristic spongiform changes in brain tissue.
Genetic Testing: In suspected cases of familial or genetic CJD, genetic testing may be used to identify mutations associated with the disease.
Exclusion of Other Conditions: Because CJD symptoms can mimic those of other neurological disorders, it’s essential to rule out alternative diagnoses through comprehensive testing and evaluation.
Treatment of Creutzfeldt Jakob Disease:
Currently, there is no cure for Creutzfeldt Jakob Disease (CJD) and no treatment that can stop or reverse its progression. Management focuses primarily on alleviating symptoms and providing supportive care to improve the patient’s quality of life. Here are the main aspects of managing CJD.
- Advertisement -
Symptomatic Treatment: Medications may be prescribed to manage symptoms such as agitation, anxiety, muscle spasms, and sleep disturbances. These treatments aim to provide comfort and reduce distress.
Nutritional Support: As swallowing difficulties (dysphagia) are common in advanced stages of CJD, feeding tubes may be necessary to ensure adequate nutrition and hydration.
Physical Therapy: Physical therapists can help maintain mobility, prevent muscle contractures, and assist with activities of daily living as the disease progresses.
Psychological Support: CJD profoundly affects cognitive function and behavior, so psychological support for both patients and their families is crucial. Counseling and support groups can help manage emotional and psychological challenges.
Palliative Care: As CJD is ultimately fatal, palliative care focuses on relieving symptoms, improving comfort, and supporting patients and families through end-of-life care decisions.
Research and Clinical Trials: Experimental treatments and clinical trials aimed at slowing disease progression or targeting the underlying prion pathology are under investigation. However, these are in early stages, and their efficacy remains uncertain.
What is Prion Diseases:
Prions are infectious agents composed solely of misfolded proteins. Unlike typical pathogens such as viruses or bacteria, prions do not contain genetic material like DNA or RNA. Instead, they arise when a normal cellular protein, called the prion protein (PrP), undergoes a misfolding process. This misfolded form, known as PrPSc (scrapie form), has the ability to convert normal PrP proteins into the abnormal form by inducing them to misfold. This conversion process leads to the accumulation of PrPSc in the brain, where it forms aggregates that damage nerve cells and cause neurodegenerative diseases known as prion diseases.
How Prions Cause Disease:
Prions cause disease through a process that involves the misfolding and accumulation of the normal prion protein (PrP) into an abnormal form, known as PrPSc (scrapie form). Here’s a step-by-step explanation of how prions lead to neurodegenerative diseases.
- Advertisement -
Misfolding of Prion Protein (PrP): Prion diseases begin when the normally occurring PrP, found on the surface of many cells, undergoes a spontaneous or induced misfolding event. This misfolded PrP, PrPSc, has a different three-dimensional structure compared to the normal PrP.
Conversion and Propagation: PrPSc acts as a template to induce the misfolding of normal PrP proteins into the abnormal PrPSc form. This conversion process is self-propagating, meaning it can perpetuate itself by converting more normal PrP proteins into PrPSc.
Accumulation and Aggregation: The accumulation of PrPSc in the brain leads to the formation of insoluble aggregates and amyloid plaques. These aggregates are resistant to normal cellular processes that break down proteins, causing them to accumulate over time.
Neurodegeneration: The accumulation of PrPSc aggregates and plaques disrupts normal cellular function in the brain. This disruption leads to neuronal damage, loss of nerve cells, and the formation of microscopic holes (spongiform changes) in brain tissue.
Clinical Manifestations: As neurodegeneration progresses, affected individuals develop a range of symptoms depending on the specific prion disease. These may include rapid cognitive decline, dementia, movement disorders, behavioral changes, and eventually severe disability.
Transmission: Prions can spread within and between species through ingestion of contaminated tissues, transplantation of infected tissues, or exposure to prion-contaminated materials. This ability to transmit prion diseases contributes to their infectious nature.
Pathological Confirmation: Diagnosis of prion diseases often involves detecting characteristic markers such as elevated levels of specific proteins (like 14-3-3 and tau) in cerebrospinal fluid, abnormal brain wave patterns on electroencephalogram (EEG), and spongiform changes observed on brain tissue examination.
FAQs about Creutzfeldt Jakob Disease:
A1: Creutzfeldt Jakob Disease (CJD) is a rare and fatal neurodegenerative disorder characterized by rapid mental deterioration and severe neurological symptoms. It belongs to a group of conditions known as transmissible spongiform encephalopathies (TSEs), caused by abnormal prion proteins that damage brain tissue. There are several forms of CJD, including sporadic, hereditary, and acquired types.
- Advertisement -
A2: Diagnosing CJD involves clinical assessment, cerebrospinal fluid (CSF) analysis, electroencephalogram (EEG), and MRI brain imaging. Genetic testing may be used for hereditary cases. A definitive diagnosis often requires a brain biopsy or autopsy, revealing prion protein accumulation and characteristic spongiform changes in brain tissue.
A3: Symptoms of CJD include cognitive decline, behavioral changes, muscle coordination problems, visual disturbances, sleep disturbances, physical symptoms like muscle stiffness and tremors, severe dementia, and end-stage symptoms leading to profound disability. The disease progresses rapidly, often leading to death within a year of onset.
A4: CJD is caused by abnormal prion proteins that lead to brain damage. The disease can occur spontaneously (sporadic CJD), be inherited through genetic mutations (hereditary CJD), or be acquired through exposure to infectious prions (acquired CJD), such as through contaminated medical procedures or consumption of beef from cattle with BSE (variant CJD).
A5: Currently, there is no cure for CJD. Treatment focuses on alleviating symptoms and providing supportive care to improve the patient’s quality of life. Experimental therapies and clinical trials are ongoing, but their efficacy remains uncertain. Symptomatic treatments include pain management, muscle relaxants, and psychological support, along with nutritional and physical therapy to maintain mobility and comfort.
-Please remember, to always consult with healthcare professionals or Doctors for personalized advice related to medical conditions.
- Advertisement -
Conclusion:
Creutzfeldt Jakob Disease (CJD) is a rare, devastating neurodegenerative disorder that significantly impacts affected individuals and their families. Characterized by rapid mental and physical deterioration, CJD belongs to a group of conditions known as transmissible spongiform encephalopathies (TSEs) and is caused by misfolded prion proteins. The disease manifests in various forms—sporadic, hereditary, and acquired—each with distinct origins and mechanisms of transmission. Despite the lack of a cure, ongoing research aims to improve our understanding of CJD, enhance diagnostic methods, and develop potential treatments. Increased awareness and continued scientific investigation are crucial to addressing the challenges posed by this fatal disease and ultimately finding ways to prevent and treat it.


